Aluminium w glinkach kosmetycznych
O tą rzecz pytano mnie już kilka razy, dlatego warto chyba odpowiedzieć publicznie.

Glina to formalnie rzecz biorąc, skała osadowa. Bardzo miękka a w stanie wilgotnym dająca się formować w rękach, ale skała. Aby osad mógł być zaliczony do glin, musi zawierać odpowiednio dużą ilość minerałów ilastych, przemieszanych z drobnym pyłem kwarcowym i skaleniowym. Tlenki żelaza i manganu nadają glinom kolor od żółtawego, przez rdzawoczerwony, brunatny, szary do ziemisto-zielonego. Glinki z małą ilością metali mogą być też białe.
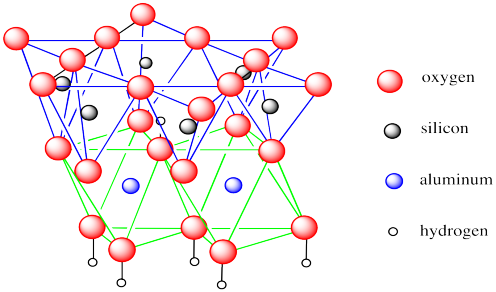
Minerały ilaste, uwalniane ze skał podczas wietrzenia, wypłukiwane w formie najdrobniejszego mułu czy wywiewane przez wiatr jako kurz, stanowią grupę krzemianów warstwowych, składających się z warstw połączonych tetraedrów tlenku krzemu SiO4 i tlenku glinu AlO4. Tlenek glinu jest izomorficzny z tlenkiem krzemu i może go naśladować, włączając się w krzemianową strukturę bez zmian. Poszczególne warstwy glinokrzemianowe są połączone wiązaniami wodorowymi, częściowo jonowymi, i potrafią wchłaniać między siebie inne substancje organiczne i nieorganiczne. Duży stopień uwodnienia powoduje, że warstwy mogą się względem siebie przesuwać, przez co glina staje się plastyczna. Właśnie zawartość tego pierwiastka w glinie stała się podstawą do zastąpienia w polskiej nomenklaturze łacińskiego aluminium nazwą glin.
Struktura warstwy kaolinu:
A jak jest z tym aluminium?
Aby glin mógł być uwalniany z gliny, powinien być rozpuszczalny w wodzie. A gliny nie są. Nie dość, że zupełnie nierozpuszczalny w wodzie jest tlenek glinu, to w glinokrzemianach dodatkowo stabilizuje go włączenie w sieć krzemianową
Kwestia ta była zresztą szczegółowo badana, zwłaszcza z uwagi na używanie ceramiki jako naczyń kuchennych, oraz używanie glinokrzemianów jako dodatków do żywności. Wydaje się, że w warunkach odczynu obojętnego uwalnianie glinu nie zachodzi. Dopiero warunki kwaśne są w stanie uwolnić część jonów. W pewnym badaniu stwierdzono dla kwasowości pH 3 (sok pomarańczowy) minerały uwalniały dość niskie poziomy aluminium, ale tylko z bardzo cienkiej (3,5 mikrometra) warstwy powierzchniowej, co sugeruje, że w przypadku naczyń ceramicznych po pewnym czasie powstanie wypłukana warstewka powstrzymująca dalsze uwalnianie. Najsłabsze wymywanie stwierdzono dla kaolinu i montmorylonitu[1]. Uwolnienie ilości mających jakieś znaczenie toksykologiczne, zachodzi dopiero pod wpływem mocniejszych kwasów.
Glinki kosmetyczne nie mają tak kwaśnego odczynu, więc uwalniania aluminium nie ma co się obawiać.W dodatku czyste glinki, przepłukane dla wymycia zanieczyszczeń, jak to zwykle jest z glinkami kosmetycznymi, mają raczej skłonność do pochłaniania rozpuszczalnych jonów glinu niż do uwalniania.
W obszernym raporcie na temat bezpieczeństwa glinokrzemianów i tlenków glinu używanych w kosmetyce, nie stwierdzono negatywnych skutków dla minerałów ilastych. [2]
Podobne pytanie dotyczyło spożywania glinek na odtruwanie - tutaj także nie ma niebezpieczeństwa, kwasy żołądkowe są bowiem dosyć rozcieńczone. Jedynym istotnym wpływem jaki stwierdzono, jest zmniejszone wchłanianie żelaza u osób zażywających glinę, bowiem absorbowała żelazo z treści żołądkowej.
Aluminium w szczepionkach
Ze względu na popularny post o ałunie jestem chyba brany za eksperta od aluminium, skoro dotyczy tego koleje pytanie zadawane mi dwa razy.
Jak wiadomo, w niektórych szczepionkach dziecięcych pojawiają się związki aluminium. Podobno zawartość w jednej nie przekracza norm bezpieczeństwa. A co jakby zsumować zawartość we wszystkich - czy wtedy mogłoby zajść jakieś niebezpieczeństwo?
Wodorotlenek glinu pojawia się w szczepionkach jako nośnik białek lub polisacharydów mających wywołać reakcję organizmu. Normy z reguły uważają za najwyższą dopuszczalną dawkę glinu 60 mg/ kg. masy ciała dziennie. Z badań na zwierzętach wynika też, że najniższe dawki mające negatywny wpływ na płody i rozwijające się młode (u szczurów) to 45 mg/kg masy ciała dziennie.
Spośród szczepionek w kalendarzu szczepień, glin zawierają:
- EUVAX B 0,25 mg w jednej dawce lub ENGERIX B też 0,25 mg w jednej dawce (stosowane wymiennie)
- DTP 0,7 mg na dawkę
-IPV 0,5 mg na dawkę
Jak łatwo zauważyć, całkowita zawartość w jednej dawce jest kilkadziesiąt razy mniejsza od najniższych dawek toksycznych. Nawet gdyby wstrzyknąć wszystkie wymienione w ciągu jednego dnia, to nie doszłoby do przekroczenia choćby dziesiątej części toksycznej dawki.
Toksyczność ałunu
Czytelnik pytał o bezpieczeństwo ałunu dla dzieci. Chodziło mu o sytuację gdyby dziecko znalazło kawałek ałunu z pękniętego sztyftu i polizało lub połknęło.
Ałun potasowy i amonowy mają działanie drażniące na błony śluzowe, dlatego pierwszą reakcją w razie połknięcia będą zapewne wymioty. Po polizaniu dziecko pewnie straci ochotę na kolejny liz, ałun ma bowiem bardzo cierpki, nieprzyjemny smak i działanie ściągające. W razie połknięcia najlepiej dać dziecku dużo wody do wypicia i skontaktować się z lekarzem. W razie polizania też dać wody, ale lekarz pewnie nie będzie potrzebny.
Toksyczność ostra związku jest niska - dawka śmiertelna to 6 g/kg masy ciała
Co takiego rozpuszcza DMSO?
Pewien dociekliwy czytelnik zainteresowany medycznymi zastosowaniami DMSO dopytywał mnie o kwestie tego co się w tym rozpuszcza, a co nie i czy ma on zastosowania medyczne.

Dimetylosulfotlenek, czyli w skrócie DMSO to cenny rozpuszczalnik, używany w medycynie i chemii organicznej. Wykazuje dużą skłonność do wnikania w tkanki organizmu, na tyle, że trzeba uważać przy pracy z nim bo po wchłonięciu metabolizuje do związków o zapachu czosnku. Ponieważ rozluźnia strukturę lipidową skóry, może ułatwiać wchłonięcie przezskórnie różnych substancji, w tym wielu leków, dlatego czyni się próby z wykorzystaniem do terapii bez konieczności zastrzyków. Sam w sobie ma zresztą działanie przeciwzapalne.
Bywa rozpuszczalnikiem środków na grzybicę stóp, które zwykle dość słabo przenikają przez zrogowaciały naskórek na podeszwach, może być też składnikiem maści przeciwbólowych do działania miejscowego, może rozmiękczać skórę przy twardzinie i przerastających bliznach. Robiono na ten temat badania [3]
Drugie pytanie dotyczyło możliwości rozpuszczania chondroityny przez DMSO i smarowania tym stawów jako alternatywy dla suplementacji doustnej.
Chondroityna to mukopolisacharyd będący składnikiem chrząstek stawowych i w formie siarczanu stanowiący wraz z glukozaminą jeden z czynników zapewniających lepszy poślizg. W związku z tym powstał pomysł, że jeśli zbyt mała ilość mazi stawowej i mała elastyczność chrząstek stawu powodują bóle, to zażywanie chondroityny i dostarczanie jej do organizmu, powinno leczyć bądź powstrzymywać rozwój schorzeń stawów poprzez dostarczenie organizmowi składnika do wytworzenia większej ilości mazi. Na tym też opiera się potężna gałąź przemysłu suplemenciarskiego.
Niestety w niedawno opublikowanej potężnej metaanalizie wielu badań wykazano, że w porównaniu z placebo zażywanie chondroityny lub glukozaminy lub obu tych środków razem, nie zmniejsza bólów ani innych objawów[4]
Wygląda na to, że samo dostarczenie organizmowi składników potrzebnych do wytworzenia czegośtam w którymś organie, wcale nie musi go pobudzać aby tą substancję jednak wytworzyć, zwłaszcza że niedostateczne wydzielanie tej substancji wcale nie musi być wynikiem niedoboru podstawowego składnika, może być skutkiem innych czynników, w tym genetycznych. Dlatego jeśli przyjrzycie się emitowanym obecnie reklamom takich pigułek, zauważycie że wcale nie stwierdzają one wprost, iż zażycie tego składnika powstrzyma chorobę. Zamiast tego zawierają ogólniki, typu "glukozamina jest składnikiem chrząstki stawowej" i "maź stawowa ma decydujące znaczenie w zapewnieniu elastyczności stawu" i na koniec, że ich pigułka zawiera podwójną czy potrójną dawkę, zaś konstrukcję logiczną "skoro to jest składnikiem chrząstki a chrząstka jest potrzebna w stawach, to pomoże mi na stawy" widz musi sam sobie przeprowadzać, w czym udanie pomaga mu sugestia pokazywanego w reklamie pana którego bolą stawy, który bierze ich tabletki a potem wygląda na mniej cierpiącego.
Ale wróćmy do pytania - czy chondroityna rozpuszcza się w DMSO? Cóż - wedle obszernego podsumowania rozpuszczalności różnych substancji w tym rozpuszczalniku, chondroityna jest nie rozpuszczalna. Słabo rozpuszcza się chlorowodorek glukozaminy, natomiast bardzo dobrze przeciwbólowy Ibuprofen.[5]
Tak że raczej nie tędy droga.
Gdzie zbadać wodę?
Miałem też pytania o to gdzie można samemu zbadać wodę ze studni, źródła czy ujęcia.
Z tego co się orientuję, takie badania oferują jednostki Sanepidu, zbadają one zawartość metali ciężkich, czystość bakteriologiczną i własności fizyczne, jak zamulenie, obecność kwasów humusowych, zażelazienie itp. Oczywiście są to badania płatne.
Próbkę wody może pobrać wysłany pracownik, ale można ją też pobrać samemu. Warszawska stacja podaje na swojej stronie specyfikację sposobu pobierania próbek, aby nadawały się do badania.[6]
Dezodorant z podbiałem
Świadoma konsumentka zadała mi pytanie w sprawie używanego dezodorantu, który zawierał w swoim składzie podbiał pospolity. Chcąc dowiedzieć się czegoś o tym składniku, dowiedziała się że roślina zawiera uszkadzające wątrobę alkaloidy. I stąd powstało pytanie, czy taki alkaloid może wchłaniać się przez skórę?
Podbiał to cenne zioło lecznicze, lecz dopiero w ostatnich latach zorientowano się, że może zawierać alkaloidy pirolizydynowe, o działaniu hepatotoksycznym, które w wyniku zażywania dłuższym niż dwa miesiące mogą powodować toksyczną niewydolność wątroby. Ich zawartość w surowcach zbieranych w Europie jest jednak bardzo niska - w polskim badaniu stwierdzono poziomy rzędu 0,0013%[7]

W jednym badaniu na szczurach stwierdzono minimalne wchłanianie przezskórne alkaloidów żywokostu, w ilości 20 razy mniejszej niż przy podaniu doustnym. Stwierdzono też, że przy chłonięciu przeskórnym, nie dochodzi do zamiany nietoksycznego N-tlenku w toksyczną formę zredukowaną, co zwykle następowało w jelitach po spożyciu.[8] Zatem wydaje się, że kosmetyków z roślin zawierających te alkaloidy można używać bezpiecznie.
Czy to cyjanek?
Dwa lata temu czytelnik przesłał mi zdjęcie białego proszku, z pytaniem czy to cyjanek potasu. Cóż mogłem odpowiedzieć... Spektroskopu w oczach nie mam. Odpisałem, że może to być cyjanek albo soda, albo sól morska, albo cokolwiek innego bo bardzo wiele soli tak wygląda. Nie wiem o co chodziło.
----------
[1] http://www.clays.org/journal/archive/volume%2026/26-6-434.pdf
[2] http://www.ncbi.nlm.nih.gov/pubmed/12851164
[3] http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3460663/
[4] http://www.bmj.com/content/341/bmj.c4675
[5] http://www.gaylordchemical.com/uploads/images/pdfs/literature/102B_english.pdf
[6] http://www.wsse.waw.pl/PageContent.aspx?SubMenuID=100
[7] https://depot.ceon.pl/bitstream/handle/123456789/1121/hp%2058%204%202012%2062.pdf?sequence=1&isAllowed=y
[8] http://www.ncbi.nlm.nih.gov/pubmed/7128756?dopt=Abstract